Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain
the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in
Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles
and JavaScript.
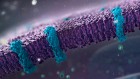
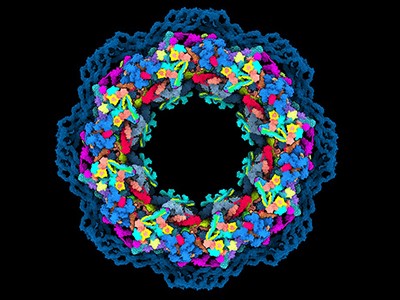
A top-down view of the human nuclear pore complex, the largest molecular machine in human cells. Credit: Agnieszka Obarska-Kosinska
For more than a decade, molecular biologist Martin Beck and his colleagues have been trying to piece together one of the world’s hardest jigsaw puzzles: a detailed model of the largest molecular machine in human cells.
This behemoth, called the nuclear pore complex, controls the flow of molecules in and out of the nucleus of the cell, where the genome sits. Hundreds of these complexes exist in every cell. Each is made up of more than 1,000 proteins that together form rings around a hole through the nuclear membrane.
These 1,000 puzzle pieces are drawn from more than 30 protein building blocks that interlace in myriad ways. Making the puzzle even harder, the experimentally determined 3D shapes of these building blocks are a potpourri of structures gathered from many species, so don’t always mesh together well. And the picture on the puzzle’s box — a low-resolution 3D view of the nuclear pore complex — lacks sufficient detail to know how many of the pieces precisely fit together.
In 2016, a team led by Beck, who is based at the Max Planck Institute of Biophysics (MPIBP) in Frankfurt, Germany, reported a model1 that covered about 30% of the nuclear pore complex and around half of the 30 building blocks, called Nup proteins.
Then, last July, London-based firm DeepMind, part of Alphabet — Google’s parent company — made public an artificial intelligence (AI) tool called AlphaFold2. The software could predict the 3D shape of proteins from their genetic sequence with, for the most part, pinpoint accuracy. This transformed Beck’s task, and the studies of thousands of other biologists (see ‘AlphaFold mania’).
“AlphaFold changes the game,” says Beck. “This is like an earthquake. You can see it everywhere,” says Ora Schueler-Furman, a computational structural biologist at the Hebrew University of Jerusalem in Israel, who is using AlphaFold to model protein interactions. “There is before July and after.”
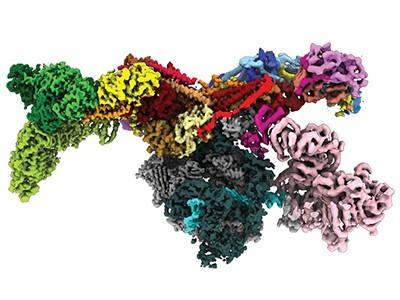
Using AlphaFold, Beck and others at the MPIBP — molecular biologist Agnieszka Obarska-Kosinska and a group led by biophysicist Gerhard Hummer — as well as a team led by structural modeller Jan Kosinski, at the European Molecular Biology Laboratory (EMBL) in Hamburg in Germany, could predict shapes for human versions of the Nup proteins more accurately. And by taking advantage of a tweak that helped AlphaFold to model how proteins interact, they managed to publish a model last October that covered 60% of the complex3. It reveals how the complex stabilizes holes in the nucleus, as well as hinting at how the complex controls what gets in and out.
In the past half-year, AlphaFold mania has gripped the life sciences. “Every meeting I’m in, people are saying ‘why not use AlphaFold?’,” says Christine Orengo, a computational biologist at University College London.
In some cases, the AI has saved scientists time; in others it has made possible research that was previously inconceivable or wildly impractical. It has limitations, and some scientists are finding its predictions to be too unreliable for their work. But the pace of experimentation is frenetic.
Even those who developed the software are struggling to keep up with its use in areas ranging from drug discovery and protein design to the origins of complex life. “I wake up and type AlphaFold into Twitter,” says John Jumper, who leads the AlphaFold team at DeepMind. “It’s quite the experience to see everything.”
A startling success
AlphaFold caused a sensation in December 2020, when it dominated a contest called the Critical Assessment of Protein Structure Prediction, or CASP. The competition, held every two years, measures progress in one of biology’s grandest challenges: determining the 3D shapes of proteins from their amino-acid sequence alone. Computer-software entries are judged against structures of the same proteins determined using experimental methods such as X-ray crystallography or cryo-electron microscopy (cryo-EM), which fire X-rays or electron beams at proteins to build up a picture of their shape.
The 2020 version of AlphaFold was the software’s second edition. It had also won the 2018 CASP, but its earlier efforts mostly weren’t good enough to stand in for experimentally determined structures, says Jumper. However, AlphaFold2’s predictions were, on average, on par with the empirical structures.
It wasn’t clear when DeepMind would make the software or its predictions widely available, so researchers used information from a public talk by Jumper, and their own insights, to develop their own AI tool, called RoseTTAFold.
Then on 15 July 2021, papers describing RoseTTAFold and AlphaFold2 appeared2,4, along with freely available, open-source code and other information needed for specialists to run their own versions of the tools. A week later, DeepMind announced that it had used AlphaFold to predict the structure of nearly every protein made by humans, as well as the entire ‘proteomes’ of 20 other widely studied organisms, such as mice and the bacterium Escherichia coli — more than 365,000 structures in total (see ‘What’s known about proteomes’). DeepMind also publicly released these to a database maintained by the EMBL’s European Bioinformatics Institute (EMBL–EBI), in Hinxton, UK. That database has since swelled to almost one million structures.
Source: E. Porta-Pardo et al. PLoS Comput. Biol.18, e1009818 (2022).
This year, DeepMind plans to release a total of more than 100 million structure predictions. That is nearly half of all known proteins — and hundreds of times more than the number of experimentally determined proteins in the Protein Data Bank (PDB) structure repository.
AlphaFold deploys deep-learning neural networks: computational architectures inspired by the brain’s neural wiring to discern patterns in data. It has been trained on hundreds of thousands of experimentally determined protein structures and sequences in the PDB and other databases. Faced with a new sequence, it first looks for related sequences in databases, which can identify amino acids that have tended to evolve together, suggesting they’re close in 3D space. The structure of existing related proteins provides another way to estimate distances between amino-acid pairs in the new sequence.
AlphaFold iterates clues from these parallel tracks back and forth as it tries to model the 3D positions of amino acids, continually updating its estimate. Specialists say the software’s application of new ideas in machine learning research seems to be what makes AlphaFold so good — in particular, its use of an AI mechanism termed ‘attention’ to determine which amino-acid connections are most salient for its task at any moment.
The network’s reliance on information about related protein sequences means that AlphaFold has some limitations. It is not designed to predict the effect of mutations, such as those that cause disease, on a protein’s shape. Nor was it trained to determine how proteins change shape in the presence of other interacting proteins, or molecules such as drugs. But its models come with scores that gauge the network’s confidence in its prediction for each amino-acid unit of a protein — and researchers are tweaking AlphaFold’s code to expand its capabilities.
By now, more than 400,000 people have used the EMBL-EBI’s AlphaFold database, according to DeepMind. There are also AlphaFold ‘power users’: researchers who’ve set up the software on their own servers or turned to cloud-based versions of AlphaFold to predict structures not in the EMBL-EBI database, or to dream up new uses for the tool.
Solving structures
Biologists are already impressed with AlphaFold’s ability to solve structures. “Based on what I’ve seen so far, I trust AlphaFold quite a lot,” says Thomas Boesen, a structural biologist at Aarhus University in Denmark. The software has successfully predicted the shapes of proteins that Boesen’s centre has determined but not yet published. “That’s a big validation from my side,” he says. He and Aarhus microbial ecologist Tina Šantl-Temkiv are using AlphaFold to model the structure of bacterial proteins that promote the formation of ice — and which could contribute to the cooling effects of ice in clouds — because biologists haven’t been able to fully determine the structures experimentally5.
As long as a protein curls up into a single well-defined 3D shape — and not all do — AlphaFold’s prediction can be hard to beat, says Arne Elofsson, a protein bioinformatician at Stockholm University. “It’s a one-click solution to get probably the best model you’re going to get.”
Where AlphaFold is less confident, “it’s very good at telling you when it doesn’t work”, Elofsson says. In such cases, predicted structures can resemble floating spaghetti strands (see ‘The good, the bad and the ugly’). This often corresponds to regions of proteins that lack a defined shape, at least in isolation. Such intrinsically disordered regions — which make up around one-third of the human proteome — might become well defined only when another molecule, such as a signalling partner, is present.
Images: J. M. Thornton et al. Nature Med.27, 1666–1669 (2021).
Norman Davey, a computational biologist at the Institute of Cancer Research in London, says AlphaFold’s ability to identify disorder has been a game-changer for his work studying the properties of these regions. “Instantly there was a huge increase in the quality of the predictions we had, without any effort on our part,” he says.
AlphaFold’s dump of protein structures into the EMBL-EBI database is also immediately being put to use. Orengo’s team is searching it to identify fresh kinds of proteins (without experimentally verifying them) and has turned up hundreds, perhaps thousands, of potentially new protein families, expanding scientists’ knowledge of what proteins look like and can do. In another effort, the team is scouring databases of DNA sequences harvested from the ocean and waste water, to try to identify new plastic-eating enzymes. Using AlphaFold to quickly approximate the structures of thousands of proteins, the researchers hope to better understand how enzymes evolved to break down plastic, and potentially to improve them.
The ability to transform any protein-coding gene sequence into a reliable structure should be especially powerful for evolution studies, says Sergey Ovchinnikov, an evolutionary biologist at Harvard University in Cambridge, Massachusetts. Researchers compare genetic sequences to determine how organisms and their genes are related across species. For distantly related genes, comparisons might fail to turn up evolutionary relatives because the sequences have changed so much. But by comparing protein structures — which tend to change less rapidly than genetic sequences — researchers might be able to uncover overlooked ancient relationships. “This opens up an amazing opportunity to study the evolution of proteins and the origins of life,” says Pedro Beltrao, a computational biologist at the Swiss Federal Institute of Technology in Zurich.
To test this idea, a team led by Martin Steinegger, a computational biologist at Seoul National University, and his colleagues used a tool they developed, called Foldseek, to look for relatives of the RNA-copying enzyme of SARS-CoV-2 — the virus that causes COVID-19 — in the EMBL-EBI’s AlphaFold database6. This search turned up previously unidentified possible ancient relatives: proteins across eukaryotes — including slime moulds — that resemble, in their 3D structure, enzymes called reverse transcriptases that viruses such as HIV use to copy RNA into DNA, despite very little similarity at the genetic-sequence level.
Experimental assistant
For scientists who want to determine the detailed structure of a specific protein, an AlphaFold prediction isn’t necessarily an immediate solution. Rather, it provides an initial approximation that can be validated or refined by experiment — and which itself helps to make sense of experimental data. Raw data from X-ray crystallography, for instance, appear as patterns of diffracted X-rays. Typically, scientists need a starting guess at a protein’s structure to interpret these patterns. Previously, they’d often cobble together information from related proteins in the PDB or use experimental approaches, says Randy Read, a structural biologist at the University of Cambridge, UK, whose lab specialized in some of these methods. Now, AlphaFold’s predictions have rendered such approaches unnecessary for most X-ray patterns, Read says, and his lab is working to make better use of AlphaFold in experimental models. “We’ve totally refocused our research.”
He and other researchers have used AlphaFold to determine crystal structures from X-ray data that were uninterpretable without an adequate starting model. “People are solving structures that, for years, had not been solved,” says Claudia Millán Nebot, a former postdoc in Read’s lab who now works at the analytics firm SciBite in Cambridge. She expects to see a glut of new protein structures submitted to the PDB, in large part as a result of AlphaFold.
The same is true for labs specializing in cryo-EM, which captures pictures of flash-frozen proteins. In some instances, AlphaFold’s models have accurately predicted unique features of proteins called G-protein-coupled receptors (GPCRs) — which are important drug targets — that other computational tools got wrong, says Bryan Roth, a structural biologist and pharmacologist at the University of North Carolina at Chapel Hill. “It seems to be really good for generating first models, which we then refine with some experimental data,” he says. “That saves us some time.”
But Roth adds that AlphaFold isn’t always that accurate. Of the several dozen GPCR structures his lab has solved, but not yet published, he says, “about half the time, the AlphaFold structures are fairly good, and half the time they’re more or less useless for our purposes”. In some instances, he says, AlphaFold labels predictions with high confidence, but experimental structures show that it is wrong. Even when the software gets it right, it cannot model how a protein would look when bound to a drug or other small molecule (ligand), which can substantially alter the structure. Such caveats make Roth wonder how useful AlphaFold will be for drug discovery.
It’s increasingly common in drug-discovery efforts to use computational-docking software that screens billions of small molecules to find some that might bind to proteins — one indication that they could make useful drugs. Roth is now working with Brian Shoichet, a medicinal chemist at the University of California, San Francisco, to see how AlphaFold’s predictions compare with experimentally determined structures in this exercise.
Shoichet says they are limiting their work to proteins for which AlphaFold’s prediction chimes with experimental structures. But even in these instances, the docking software is turning up different drug hits for the experimental structure and AlphaFold’s take, suggesting that small discrepancies could matter. “That doesn’t mean we won’t find new ligands, we’ll just find different ones,” says Shoichet. His team is now synthesizing potential drugs identified using AlphaFold structures, and testing their activity in the lab.
Critical optimism
Researchers at pharmaceutical companies and biotechnology firms are excited about AlphaFold’s potential to help with drug discovery, says Shoichet. “Critical optimism is how I’d describe it.” In November 2021, DeepMind launched its own spin-off, Isomorphic Labs, which aims to apply AlphaFold and other AI tools to drug discovery. But the company has said little else about its plans.
Karen Akinsanya, who leads therapeutics development at Schrödinger, a drug-discovery firm headquartered in New York City that also publishes chemical-simulations software, says she and her colleagues are already having some success using AlphaFold structures, including for GPCRs, in virtual screens and compound design for drug candidates. She finds that, just as with experimental structures, extra software is needed to get at the fine details of amino-acid side chains or locations where individual hydrogen atoms might sit. Once this is done, AlphaFold structures have proved good enough to guide drug discovery — in some cases.
“It’s hard to say ‘this is a panacea’; that because you can do it very well for one structure — surprisingly and excitingly well — that it is eminently applicable to all structures. It clearly isn’t,” Akinsanya says. And she and her colleagues have found that AlphaFold’s accuracy predictions don’t show whether a structure will be useful for later drug screening. AlphaFold structures will never fully replace experimental ones in drug discovery, she says. But they might speed up the process by complementing experimental methods.
Drug developers curious about AlphaFold received good news in January, when DeepMind lifted a key restriction on its use for commercial applications. When the company released AlphaFold’s code in July 2021, it had stipulated that the parameters, or weights, needed to run the AlphaFold neural network — the end result of training the network on hundreds of thousands of protein structures and sequences — were for non-commercial use only. Akinsanya says this was a bottleneck for some in industry, and there was a “wave of excitement” when DeepMind changed tack. (RoseTTAFold came with similar restrictions, says Ovchinnikov, one of its developers. But the next version will be fully open-source.)
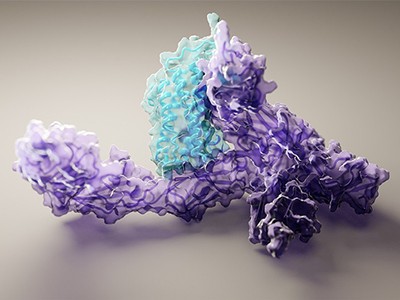
AI tools are not just changing how scientists determine what proteins look like. Some researchers are using them to make entirely new proteins. “Deep learning is completely transforming the way that protein design is being done in my group,” says David Baker, a biochemist at the University of Washington in Seattle and a leader in the field of designing proteins, as well as predicting their structures. His team, with computational chemist Minkyung Baek, led the work to develop RoseTTAFold.
Baker’s team gets AlphaFold and RoseTTAFold to “hallucinate” new proteins. The researchers have altered the AI code so that, given random sequences of amino acids, the software will optimize them until they resemble something that the neural networks recognize as a protein (see ‘Dreaming up proteins’).
Images: Ref. 7
Enjoying our latest content?
Login or create an account to continue
Access the most recent journalism from Nature's award-winning team
Explore the latest features & opinion covering groundbreaking research






 DeepMind’s AI predicts structures for a vast trove of proteins
DeepMind’s AI predicts structures for a vast trove of proteins
 DeepMind’s AI for protein structure is coming to the masses
DeepMind’s AI for protein structure is coming to the masses
 Artificial intelligence powers protein-folding predictions
Artificial intelligence powers protein-folding predictions